Photonic Materials from ab-initio Theory
Principal investigator: Prof. Dr. Wolf Gero Schmidt
Project manager/Main research: Dr. Uwe Gerstmann
Researchers: Prof. Dr. Wolf Gero Schmidt, Dr. Uwe Gerstmann
Title of research project: Photonic Materials from ab-initio Theory
Funding: Transregional Collaborative Research Center TRR 142
Project partners: Paderborn University
Project endurance: 04.2019 - 04.2022
Project area: 307-01
Cluster: Noctua Cluster at PC2
Software: Quantum ESPRESSO, VASP
Introduction:
Accurate parameter-free calculations of optical response functions for real materials and nanostructures still represent a major challenge for computational materials science. This project concentrates on the development and application of ab-initio methods which provide access to linear and nonlinear optical spectra. It explores, on the atomistic level, how the material structure, its composition and defects but also external parameters like stress, temperature or magnetic fields influence the optical response. In addition, it explores how optical excitations modify the material electronic and atomic structure as well as the time dynamics of optical excitation and de-excitation.
Methods:
The calculations start from an accurate description of the structural and electronic ground-state properties within density-functional theory (DFT). Time-dependent DFT in conjunction with a Berry-phase formulation of the dynamical polarization accounts for many body effects in the optical response in an efficient way without recourse to virtual orbitals. More precise schemes based on many body perturbation theory, such as the GW approximation for the quasiparticle energies or the Bethe-Salpeter equation (BSE) for the linear optical response, are used to benchmark the TDDFT results. Both zero-point vibrations and thermal lattice vibrations are included in the calculations. The computational methods developed in this project are applied to a wide range of II-VI, III-V and nitride semiconductors and nanostructures as well as ferroelectric materials such as potassium titanyl phosphate.
Results:
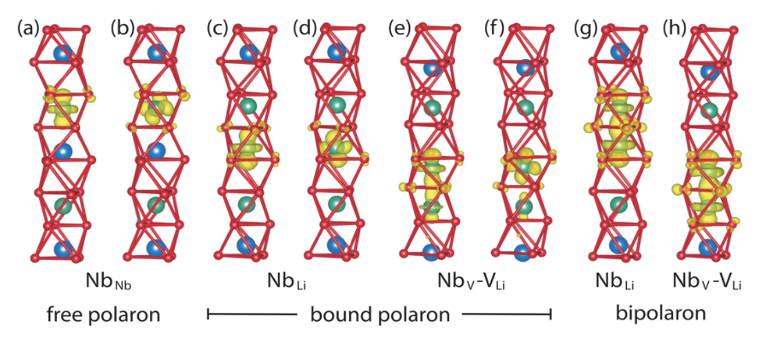
Polarons in dielectric crystals play a crucial role for applications in integrated electronics and optoelectronics. In Reference [1], we used DFT and Green’s function methods to explore the microscopic structure and spectroscopic signatures of electron polarons in lithium niobate. Total-energy calculations and the comparison of calculated electron paramagnetic resonance data with available measurements reveal the formation of bound polarons at NbLi antisite defects with a quasi-Jahn-Teller distorted, tilted configuration. The defect-formation energies further indicate that (bi)polarons may form not only at NbLi antisites but also at structures where the antisite Nb atom moves into a neighboring empty oxygen octahedron. Based on these structure models (see Fig. 1), and on the calculated charge-transition levels and potential-energy barriers, we proposed two mechanisms for the optical and thermal splitting of bipolarons, which provide a natural explanation for the reported two-path recombination of (bi)polarons. Optical-response calculations based on the Bethe-Salpeter equation, in combination with available experimental data and new measurements of the optical absorption spectrum, further corroborate the geometries proposed here for free and defect-bound (bi)polarons. The magnetic signatures of Ti3+ centers in KTP were studied within DFT in Reference [2]. The hyperfine tensor elements are very sensitive to the structural surrounding. Therefore, the paramagnetic hyperfine splittings were used to evaluate the defect models. For each of the four experimentally observed electron paramagnetic resonance (EPR) spectra, we identified a defect model that reproduces the paramagnetic signature, see, e.g., Figure 2. All of them are electron donors, whereby one specific Ti atom can be identified, whose formal oxidation number is lowered from 4+ in the ideal crystal to 3+. The related charge redistribution leads to a strong polarization of the corresponding Ti3+ center. However, in three cases a second Ti atom, connected to the first by a mutual polarized O atom, is polarized too. Positively charged O vacancies at the lattice site O(10) are unique in leading to the formation of the only Ti3+ center that is stable at room temperature. This defect induces a defect state within the band gap, which may be excited during second harmonic generation (SHG) applications and thus is a plausible candidate to explain the so-called gray tracking, i.e., photochromic damage.
Discussion:
The modeling of excited-state properties is a long-standing challenge for density-functional theory, which by definition refers to the materials ground state. It is typically tackled using many-body perturbation theory. This was also the standard approach most frequently used in our work, see, e.g., Ref. [1]. In addition, however, we found the application of constrained DFT to be very promising. It has been applied, for example, in Refs. [3,5]. In any case, the computational requirements, with respect to both CPU time as well as memory are extremely high, in particular for the quasiparticle approach and the calculation of nonlinear optical spectra.
Outlook:
The project leads to a better understanding of existing materials and contributes to the design of new photonic materials.
[1] F Schmidt, AL Kozub, T Biktagirov, C Eigner, C Silberhorn, A Schindlmayr, WG
Schmidt, U Gerstmann «Free and defect-bound (bi)polarons in LiNbO3: Atomic
structure and spectroscopic signatures from ab initio calculations» Phys. Rev.
Research 2, 043002 (2020).
[2] A Bocchini, C Eigner, S Silberhorn, WG Schmidt, U Gerstmann «Understanding gray
track formation in KTP: Ti3+ centers studied from first principles» Phys. Rev. Materials
4, 124402 (2020).
[3] M Krenz, U Gerstmann, WG Schmidt «Photochemical Ring Opening of Oxirane
Modeled by Constrained Density Functional Theory» ACS Omega 5, 24057 (2020).
[4] J Niederhausen, RW MacQueen, K Lips, H Aldahhak, WG Schmidt, U Gerstmann
«Tetracene Ultrathin Film Growth on Hydrogen-Passivated Silicon» Langmuir 36,
(2020).
[5] SV Badalov, R Wilhelm, WG Schmidt «Photocatalytic properties of graphene-supported
titania clusters from density-functional theory» J. Comput. Chem. 41, 1921 (2020).
[6] M Rosenthal, JKN Lindner, U Gerstmann, A Meier, WG Schmidt, R Wilhelm
«A photoredox catalysed Heck reaction via hole transfer from a Ru(II)-bis(terpyridine)
complex to graphene oxide» RSC Adv. 10, 42930 (2020).
[7] complex to graphene oxide» RSC Adv. 10, 42930 (2020).
T Biktagirov, WG Schmidt, U Gerstmann «Spin decontamination for magnetic dipolar
coupling calculations: Application to high-spin molecules and solid-state spin qubits»
Phys. Rev. Research 2, 022024(R) (2020).
[8] T Biktagirov and U Gerstmann, «Spin-orbit driven electrical manipulation of the zero-
field splitting in high-spin centers in solids» Phys. Rev. Research 2, 023071 (2020).
[9] C Braun, S Neufeld, U Gerstmann, S Sanna, J Plaickner, E Speiser, N Esser, W. G.
Schmidt «Vibration-Driven Self-Doping of Dangling-Bond Wires on Si(553)-Au
Surfaces» Phys. Rev. Lett. 124, 146802 (2020).