Calculation of Bulk Phase Vibrational Circular Dichroism Spectra
Principal investigator: Prof. Dr. Barbara Kirchner
Project manager/Main research: Prof. Dr. Barbara Kirchner
Researchers: Jan Blasius, Roman Elfgen, Lars Esser, Dr. Werner Reckien, Dr. Oldamur Hollóczki, Prof. Dr. Barbara Kirchner
Title of research project: Calculation of Bulk Phase Vibrational Circular Dichroism Spectra
Funding: DFG
Project partners: -
Project endurance: Since 2019
Project area: Theoretical Chemistry
Cluster: Bonna Cluster, University of Bonn
Software: CP2K, Turbomole, Peacemaker, Travis
Introduction
Vibrational circular dichroism (VCD) spectroscopy is a powerful technique for determining the three-dimensional structure of chiral molecules. Hereby, the different attenuation of left- and right-circularly polarized infrared (IR) radiation during vibrational transitions is measured. An advantage in comparison to electronic circular dichroism is that VCD does not require the presence of any dedicated chromophore. Instead, all chemical bonds serve as probe, thus making VCD spectroscopy very sensitive to conformational changes and intermolecular interactions. Based on that, VCD spectra can be used to determine both, the absolute configuration and the conformations of molecules or aggregates.
We present a method which enables the calculation of VCD spectra in the gas as well as in the bulk phase by applying a novel weighting approach based on the quantum cluster equilibrium (QCE) theory. Opposed to classical Boltzmann weights, the QCE theory allows a mixing of differently sized oligomers at the same time, thereby ensuring the inclusion of different structural motifs and interaction patterns. The obtained QCE populations, the so-called Cluster-weights, are employed for the weighting of individual cluster VCD spectra in order to calculate an overall gas or bulk phase VCD spectrum. Explicit solvation, which is necessary for an accurate description of hydrogen bonds, is intrinsically included without neglecting monomeric structures and different conformers. The cluster-weighting approach is compared to VCD spectra from ab initio molecular dynamics (AIMD) simulations.
Methods
AIMD simulations were performed with CP2K. The Turbomole program package was used for quantum chemical calculations of molecular clusters. The cluster-weighting and the analysis of trajectories from AIMD simulations were done with our in-house software Peacemaker and Travis.
Results
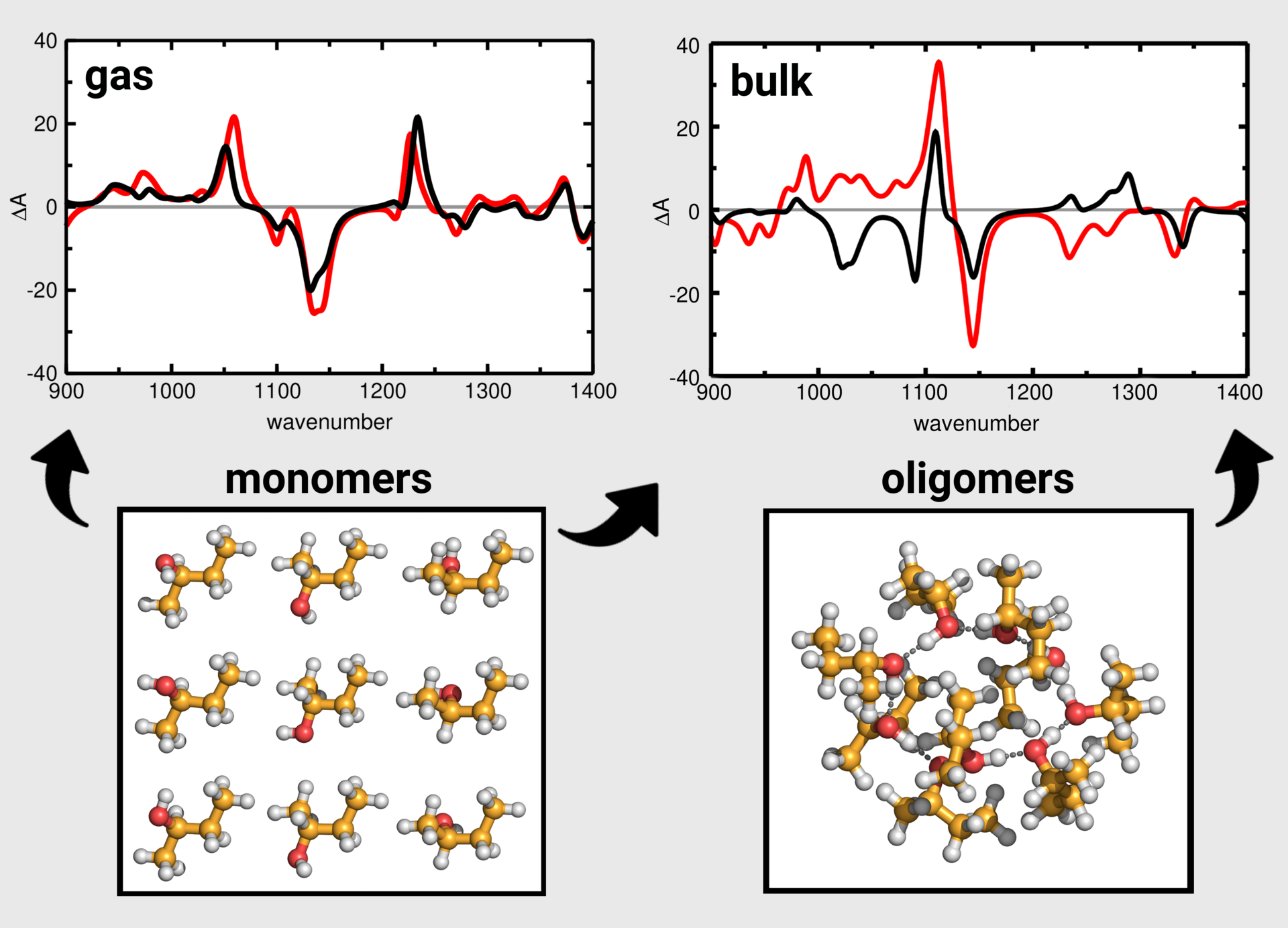
The very flexible and chiral molecule (R)-butan-2-ol was chosen to be the first test case for our novel approach.
Nine possible monomer conformers and a large quantity (several hundred) of oligomers were identified. The calculation of the VCD spectra for several cluster sets including differently sized oligomers up to decamers, showed that the inclusion of a variety of conformers for larger oligomeric clusters is not necessary and that low populated clusters can be excluded without losing accuracy. The resulting VCD spectra exhibit a very good agreement to experimental references. A slightly better match is obtained when the VCD spectra are determined from computationally much more demanding AIMD simulations which require the calculation of electric and magnetic dipole
moments in each time step. In this regard, we also considered a computationally less expensive simulation approach, in which the dipole moments are approximated by charge times position from different population analyses for partial charges. The spectra obtained by this approach are much faster to calculate and only slightly worse compared to the spectra from full simulations. Overall, all approaches allow a certain assignment of the absolute configuration of the chiral system.
In a further study, VCD spectra from AIMD simulations were calculated for three different D-glucose isomers solvated in an ionic liquid with and without water. Again we observed a very good agreement to experimental results and were able to reveal insights into intermolecular interactions.
Outlook
In further studies we want to improve the selection mechanism of the oligomers for the cluster-weighting. There are some hints that it might be possible to develop a procedure in which just the global minimum structures have to be considered. Overall, a better understanding of the meaning of cluster sets including medium sized oligomers for the accuracy of the spectra is desirable. We want to test our new protocol on other substances as well as binary systems. This would enable the calculation of concentration dependent VCD spectra in solution. Furthermore, there is a great potential in the combination of our cluster-weighting approach with a neural network ansatz.
List of Publications:
- J. Blasius, R. Elfgen, O. Hollóczki, B. Kirchner, Glucose in dry and moist ionic liquid: Vibrational circular dichroism, IR, and possible mechanisms, Phys. Chem. Chem. Phys. (2020), 22, 10726-10737. DOI: 10.1039/C9CP06798A
- J. Blasius, B. Kirchner, Cluster-weighting in Bulk Phase Vibrational Circular Dichroism, J. Phys. Chem. B (2020), 124, 7272-7283. DOI: 10.1021/acs.jpcb.0c06313
- B. Kirchner, J. Blasius, L. Esser, W. Reckien, Predicting Vibrational Spectroscopy for Flexible Molecules and Molecules with Non-Idle Environments, Adv. Theory Simul. (2020), 2000223. DOI:10.1002/adts.202000223